|
||||||

|
Introduction The term fluorescence was coined by George Gabriel Stokes (1819-1903) in his famous paper [On the Change of Refrangibility of Light, Philosophical Transactions of the Royal Society of London 142:463-562 (1852)]. It is one form of a quantum mechanical process called photoluminescence, which becomes visible as optical radiation during the relaxation of a molecule from an excited (higher energy) state to its (lower energy) ground state accompanied by the emission of a photon. Depending on the duration of the phenomenon (lifetime of the excited state), one distinguishes between fluorescence (~ 10-9...10-6 s) and phosphorescence (~ 10-3...1000 s). Principle
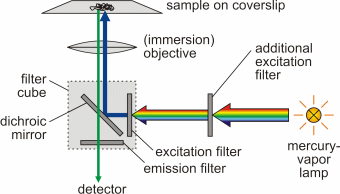
As a result of the Franck-Condon principle as well as inner non-radiative de-excitation (internal conversion), an increase of the wavelength of absorbed and emitted photon occurs (Stokes shift), Δλ = λem - λabs > 0. Fluorescence microscopy was developed by August Köhler (1866-1948) and Henry Siedentopf at Carl Zeiss AG in Jena and presented to the public on the occasion of a microscopy course at the botanical institute in Vienna in April 1908. Being a form of reflectedlight microscopy, it is more precisely called epifluorescence microscopy. It is based on a fluorescent dye (fluorophore) with which the sample or individual structures are labeled. This dye is excited by light of a certain wavelength. The light source is usually either a mercury-vapor lamp or a laser.
This technique is widely used in biology to visualize cellular components, certain proteins or other molecules of interest (examples of fluorescence images can be found in the article Cytoskeleton). With sufficiently high contrast, it is even possible to image objects whose size is far smaller than the resolution limit of light microscopes. Proper staining is of utmost importance for fluorescence microscopy. There are several methods and a myriad of markers in order to do this. Classical staining methods often require fixation of the cell, in which cellular proteins are cross-linked by application of a fixative like formaldehyde or alcohols. If dynamical behavior of a cell is of interest, the fixation approach is not practicable. In such cases a live staining method is necessary that does not significantly alter the natural physiology of the cell. Staining can be done for example by adding specifically binding fluorescently labeled ligands, adding specific antibodies (immunofluorescence) or transfection in order to make cells express marker proteins like GFP. Photobleaching Every fluorophore is prone to photobleaching, which is the natural permanent
loss of the ability of a fluorophore to fluoresce due to photochemical
destruction of the fluorophore molecules when exposed to the excitation
light. The average number of excitation-emission-cycles depends not only
on the intensity and energy of the excitation light, but also the molecular
structure of the fluorophore and its chemical environment. Some fluorophores
bleach after emission of only a few photons, while other can emit substantially
more.
Phototoxicity Phototoxicity refers to the possible effect in living cells where illuminating a fluorophore causes damage of the cells expressing it, eventually leading to selective cell death. This phenomenon is not yet fully understood, but the formation of oxygen radicals due to non-radiative energy transfer seems to be the main cause. It strongly depends on the kind of fluorescent molecule used, however it can be minimized by reducing intensity and time of illumination to avoid damage of cells during fluorescent imaging. (This article is taken from the diploma thesis of Steve Pawlizak, University of Leipzig, 2009.) |
||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||
|